生物学的解析として、Clustal W、シークエンスの生データのアライメントに適した「アライメント」、分子量の計算、 ORF の検索、塩基配列を編集しながらアミノ酸配列を見ることのできる「Amino view」、波形ファイルを表示する「Wave view」、樹形図を表示する 「Tree view」、全体像を把握するための「Over view」、 Dot blot 、 Degenerate primer の設計、 GC 含量や疎水性の程度を表す「プロット」などができます。
これらの機能を正しく使うためには「生物学」->「特性」メニューから正しい特性を選択しておく必要が有ります。DNAは金の聖杯、環状DNA はリング付きの金の聖杯、アミノ酸は銀の聖杯、その他は銅の聖杯のアイコンになります。
「生物学」->「コドン表」から「標準」または「ミトコンドリア(脊椎動物)」を選択することができます。「コドン表」は ORF の検索、「Amino view」、 Degenerate primer の設計のパラメーターとして使用されます。
| DNA 環状DNA アミノ酸 その他 | |||
Clustal W
「生物学」メニューから「Clustal W」を選択すると、Kyoto University Bioinformatics Center の Clustal W のサイトにアクセスし、全配列を対象に Clustal W のデフォルト設定でアライメントを行います。インターネットに接続している場合のみに使用可能です。
詳細な設定を行いたい場合は FASTA 形式で保存したファイルで解析できます。アライメント結果である .aln ファイルは IdentityX で読み込めます。
<参考>Clustal W、アライメント、サーチボタンの使い分けに付いて
ORF
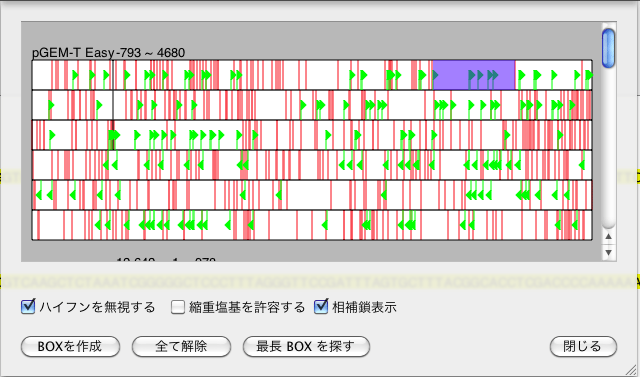
「生物学」メニューから「ORF」を選択すると、下のシートが現れます。スタートコドンは緑の旗で、ストップコドンは赤い線で表されます。クリックで ORF を選択または解除できます。「相補鎖表示」にチェックを入れると相補鎖の ORF も表示します。「最長 BOX を探す」ボタンを押すと配列の中で最長の ORF を選択します。
 |
|
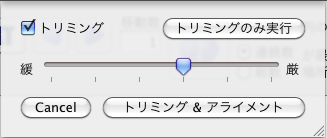
| アライメント 「生物学」メニューから「アライメント」を選択すると、右のシートが現れます。「トリミング&アライメント」ボタンを押すと配列の両端の不確定な配列をトリミングし、続けてアライメントを行います。 |
|||||
 |
|||||
| 「アライメント」は配列の向きを変えることがありますが、最上段の配列の向きは基準となり、変更されることは有りません。 「トリミングのみ実行」ボタンを押すと、トリミングされるべき範囲が選択された状態になります。トリミング範囲に問題が無ければそのままキーボードの「delete」ボタンを押して、選択範囲を削除して下さい。 「トリミング」チェックボックスのチェックを外してアライメントだけを行うことも可能です。 トリミングをやりすぎる場合やアライメントが行われない場合は条件を緩くし、トリミングが足りない場合や間違ったアライメントをする場合は条件を厳しくしてみて下さい。 <参考>Clustal W、アライメント、サーチボタンの使い分けに付いて |
|||||
|
Amino view |
|||||
 |
|||||
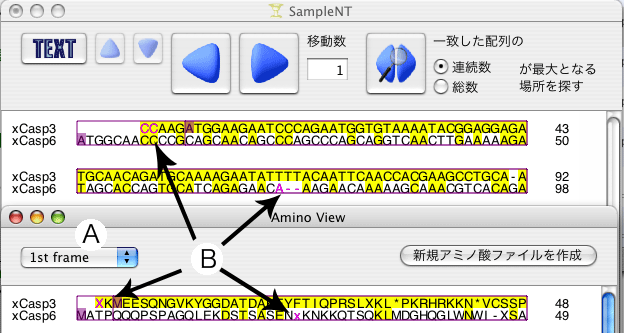
このウィンドウの左上のポップアップボタンでフレームを選択すると (A)、そのフレームでアミノ酸に変換された配列が「Amino view」ウィンドウに表示されます。選択範囲、検索結果、 BOX の位置が連動して表示されるので、対応する配列が容易に分かります。 この図は「Amino view」ウィンドウで "X" と "x" を選択したものです (B)。そのフレームに2塩基しか無い場合は "X" 、1塩基しか無い場合は "x" と表示されていることが分かります。縮重塩基を含んでいて、アミノ酸が一義的に決まらない場合や、塩基と解釈できない記号(E,F,J,L,O,P,Q,X,Zなど)が含まれる場合は "?" と表示されます。「Amino view」ウィンドウで「新規アミノ酸ファイルを作成」ボタンを押すことにより、アミノ酸配列の新規ファイルを作成できます。
Nt view
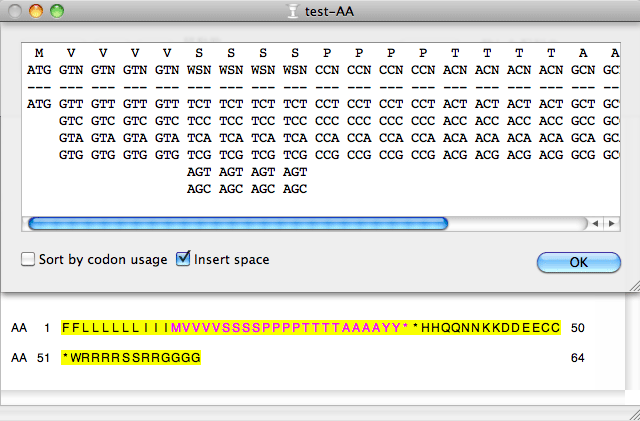
アミノ酸配列を選択し、「生物学」メニューから「Nt view」を選択すると、核酸に変換した配列を表示します。選択範囲が複数の場合は使用できません。コドン表の「Count」欄に codon usage を入力しておくと、使用頻度の高い順に表示することもできます。
 |
|
|
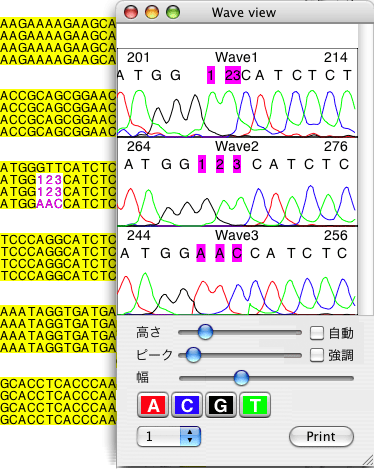
Wave view 配列は編集可能ですが、多少クセが有ります。例えば右の図で "GTT" という配列を削除してから "GG" と "CA" の間に"123"という配列をキーボードから入力したものが、 "Wave1" 、 "123" という配列をペーストしたものが "Wave2" です。 |
||||||||||
 |
||||||||||
|
"Wave1" のようにキーボードから入力した場合は始めに "1" を "G" と "C" の中間に配置し、次に "2" を "1" と "C" の中間に配置、最後に "3" を "2" と "C" の中間に配置、という手順で配置するために後ろの方が詰まってしまいます。 配列を編集してもほとんどの場合は波形データが変更されることは有りませんが、「生物学」メニューの"Rev & Comp"を使用する場合は注意が必要です。例えば上図で "GTT" という配列を "Rev & Comp" で相補鎖に変換したものが "Wave3" です。通常は配列全体に"Rev & Comp"を使用するので問題は起こりませんが、この例のように配列の一部に"Rev & Comp"を使用する場合には波形データが加工されるので、十分注意して下さい。 Wave view では表示枠の幅と高さをウィンドウ枠に合うように自動調節できます。幅は指定した列数になるように常に自動調節されます。高さは「自動」チェックボックスにチェックを入れておくと自動調節されます(右下図)。 |
||||||||||
|
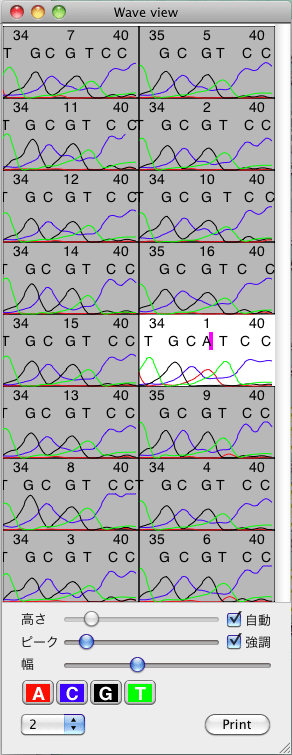
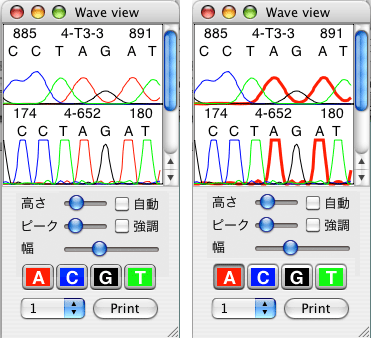
「強調」チェックボックスにチェックを入れておくと編集中の配列が強調されます(右図の配列1)。強調表示を行うとプロンプトの位置も強調されるので、編集が非常にやり易くなります。 さらに、いつでも指定した塩基の波形を強調することができるので、波形が重なっていて見にくい場合だけでなく、色盲・色弱の方などにも役立つのではないでしょうか?(下図)。左が通常の表示で、右が A を強調表示にしたものです。塩基ボタンを押す度に "強調 / 非強調" が切り替わります。 |
 |
|||||||||
 |
||||||||||
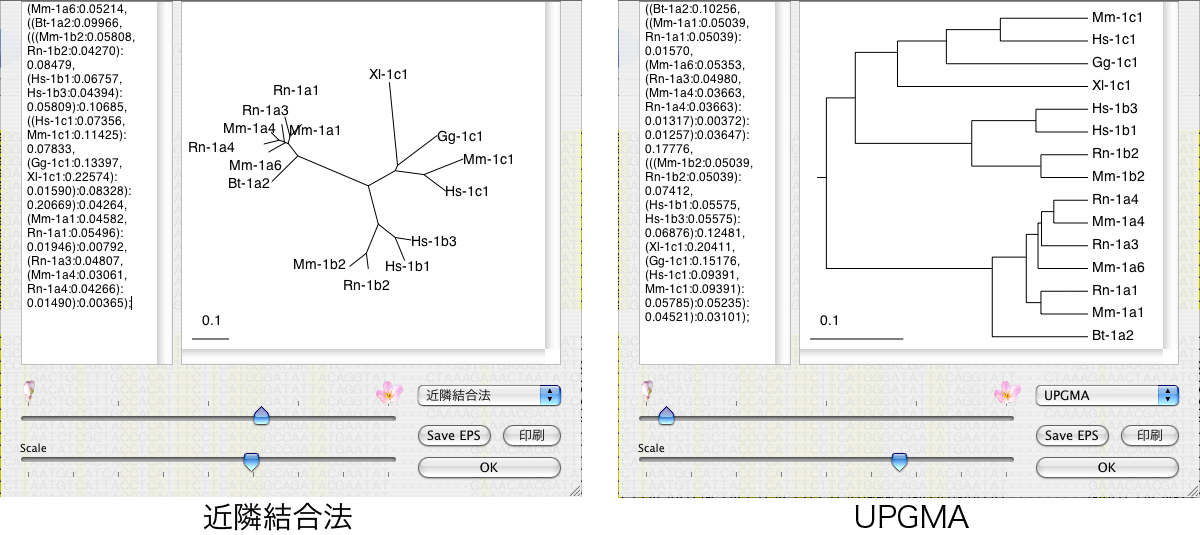
| Tree view 国立遺伝研の Clustal W のサイトのデフォルトと同じパラメーター(詳しくはこちら)で進化的距離を計算し、近隣結合法(国立遺伝研の Clustal W のサイトと同じ)と UPGMA で樹形図を作成することができます。 左のテキストフィールドには解析結果を Newick 形式で表示します。ここからコピーして他のソフトで利用することはできますが、ここに文字列をぺ−ストしても何も起こりません。 |
||||||||||
 |
||||||||||
| Over view 配列の重なり具合の全体像を見ることができます。配列を取り込んだ時点で右向きの矢印となり、配列全体に「Rev & Comp」を行うと逆向きになります。波形情報を持たない配列および向きを持たない配列は黒で表され、波形情報と向きを持つ配列は色付きで表されます。ただし、バージョン7より前のファイルはバージョン7以降で初めて開いた時に右向きの矢印となります。配列を選択し、「Change direction」ボタンを押すと向きを変えられます。この操作は「Over view」での表示が変わるだけで、配列全体に「Rev & Comp」を行うわけではありません。このシートで配列名や表示する順番を変えることができます。 ハイフンは細い線で表され、配列のある部位は太い線で表されます。太い線の太さは変更できます。 EPS 形式で保存も出来ます。 |
||||||||||
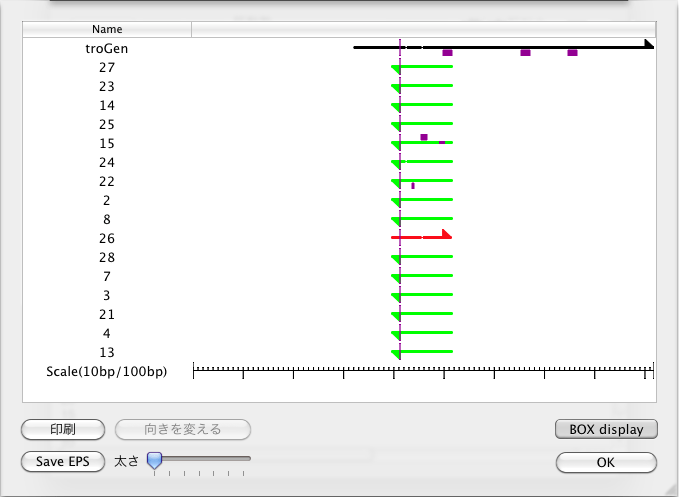 |
||||||||||
|
Dot blot |
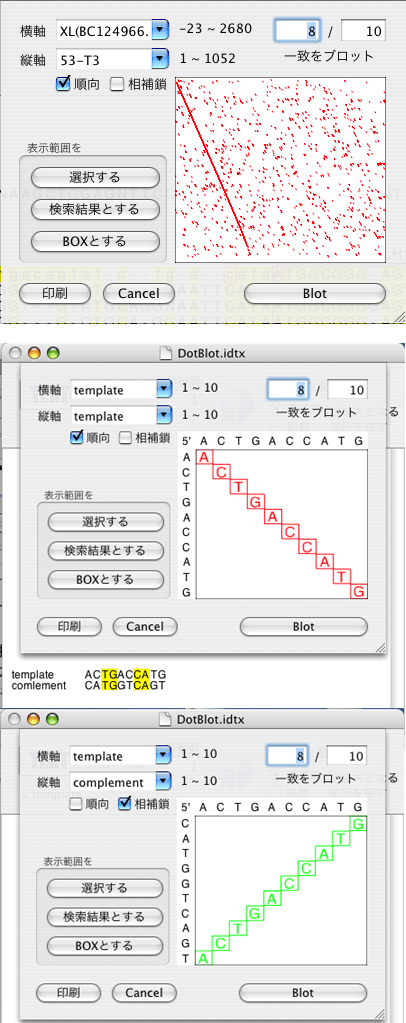 |
|||||||||
Degenerate primer の設計
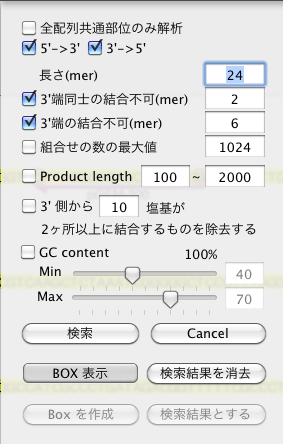
Primer の設計条件を右下の図の項目で設定します。
|
「全配列共通部位のみ解析」にチェックを入れると、解析対象が全配列共通部位のみとなり、黄色で表示されます。 |
|||
 |
|||
「組み合わせの数の最大値」とはプライマー内に縮重塩基が含まれる場合にその組み合わせの数の積の上限を指定します。例えば縮重塩基 R(A or G) が一つ含まれれば2、5つ含まれれば32 (2x2x2x2x2)、 N(A, C, G or T) が三つ含まれれば64 (4x4x4) となります。数字が小さければ小さいほど厳しい条件となり、検索されるプライマーの数は減ります。
Product length 以下の項目は通常のプライマーを設計する時に使用することを念頭にバージョン 9.0.0 で追加しました。これらの項目も使用すると degenerate primer の設計には条件が厳しすぎるかもしれません。
結果の解析 (下図は古いバージョンのものなので、実際とは多少異なります。)
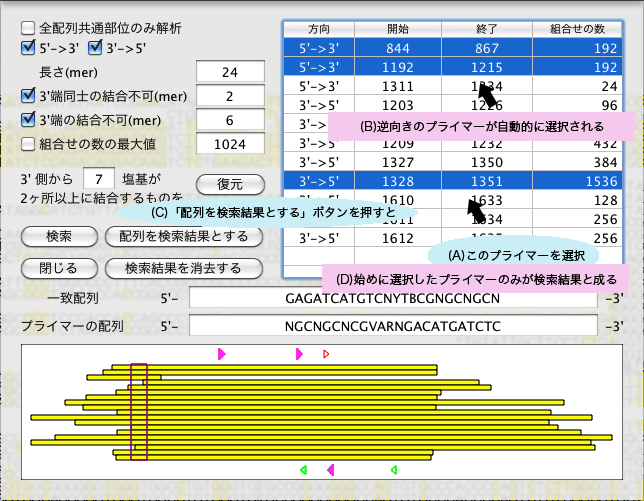
条件を満たしたプライマーがテーブルに表示されます。テーブルの項目名(「方向」など)をクリックすると、その項目の内容で並び替えを行います。テーブルの中からプライマーを選択すると、そのプライマーに相当する図の中の三角形が強調され、「一致配列」が表示されます。
「一致配列」はプライマーが一致する位置の配列を5'->3'の向きで表示し、「プライマーの配列」はプライマーの配列を5'->3'の向きで表示します。つまりプライマーの向きが5'->3'の時は同じ配列が表示され、3'->5'の時はハイブリダイズする配列が表示されることになります。
調べた配列の「特性」がアミノ酸だった場合は「一致配列」の後ろに対応するアミノ酸が括弧内に表示されます「例: ATGATGATG(MMM)」。「配列を検索結果とする」ボタン(C)で、その部位を記録できます。「BOX を作成」ボタンで順向きまたは逆向き矢印の表示属性を持った BOX を作成します。
「5'->3'」, 「3'->5'」 両方のチェックボックスにチェックを入れていた場合にテーブルからプライマーを選択すると(A)、指定した条件でそのプライマーとダイマーを作らない逆向きのプライマーが自動的に選択されます(B)。この時、テーブルでは複数のプライマーが選択された状態になっていますが、「配列を検索結果とする」ボタン(C)で検索結果に登録されるプライマーは始めに選択した一つだけです(D)。
|
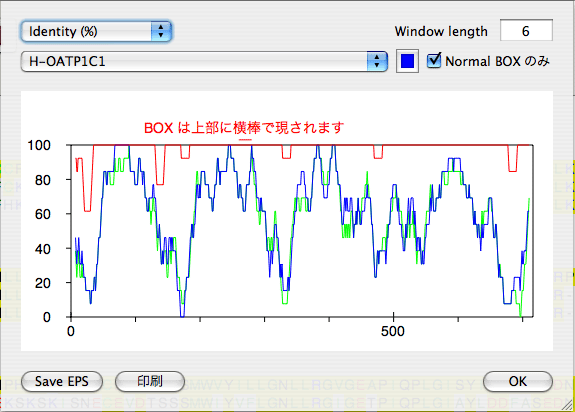
プロット |
 |
|||||||||||||||||||
|
BOX の表示は Normal BOX のみを表示するか、全ての BOX を表示するかの選択ができます。各配列ごとに色を指定することができます。この色は保存され、グラフ上部の BOX 表示の色にも使われます。 |
||||||||||||||||||||
|
||||||||||||||||||||