[bioinfo]amplicon_sorterでnanopore amplicon sequenceのデータをde novoアセンブルする
インストール方法については、上坂さんの情報に従った。
mamba create -n amplicon_sorter python=3.9 -y
conda activate amplicon_sorter
python3 -m pip install python-Levenshtein
python3 -m pip install edlib
python3 -m pip install biopython
python3 -m pip install matplotlib
#本体
git clone https://github.com/avierstr/amplicon_sorter.git
cdamplicon_sorter/Nanopore R10.4.1のフローセル上でLSK-SQK114で作ったライブラリを読む場合、下記のように結構クオリティは高い。
が、qscore > 12が推奨されているので、NanoFiltでqscore > 12でfiltorateする。pirmer annealing sitesを確認したいのでcropはしない。
cat concat.fastq | NanoFilt -q 12 > filtered_concat.fastqamplicon_sorterの手順として、まずヒストグラムでリード長の分布を確認する。ampliconの場合、目的産物のピークが存在するはずである。今回のamplicon sequencingの場合は、二つの産物を混ぜているので二つのピークが存在する。まず短い方は4.8 kbから5.6 kbあたりにピークが収まる。-minと-maxを調整して、何度かヒストグラムを描かせて赤点線がピークを包含するようにする。
python3 amplicon_sorter.py -i filtered_concat.fastq -o amplicon_sorter_out -min 4800 -max 5600 -ho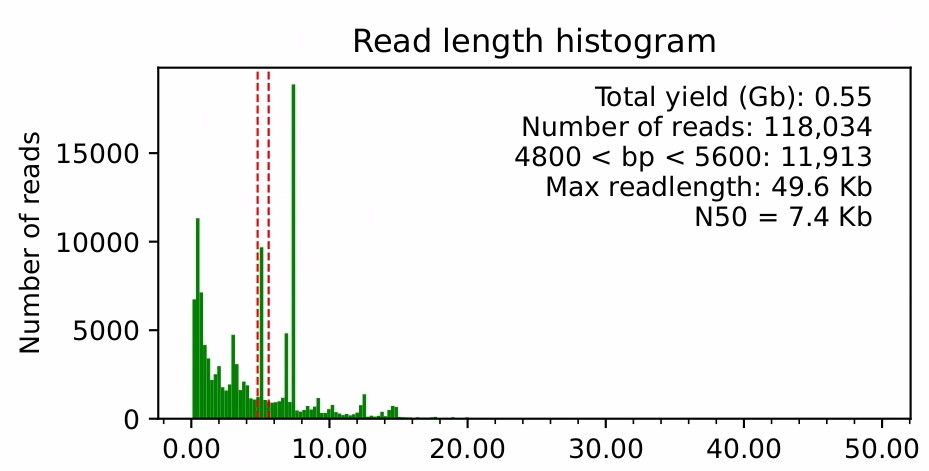
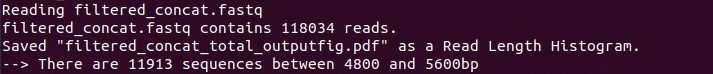
これが確認出来たら、実際にランを行う。デフォルトでは抽出するリード数が10000となっている。total evidence信奉者はこの範囲のすべてのデータを使いたいので”-maxr 20000″として、すべてのリードを使ってランを行う。”-np 20″として、core i7 12700KFをフルに回して10分程度かかる。
python3 amplicon_sorter.py -i filtered_concat.fastq -o amplicon_sorter_out -min 4800 -max 5600 -np 20 -maxr 20000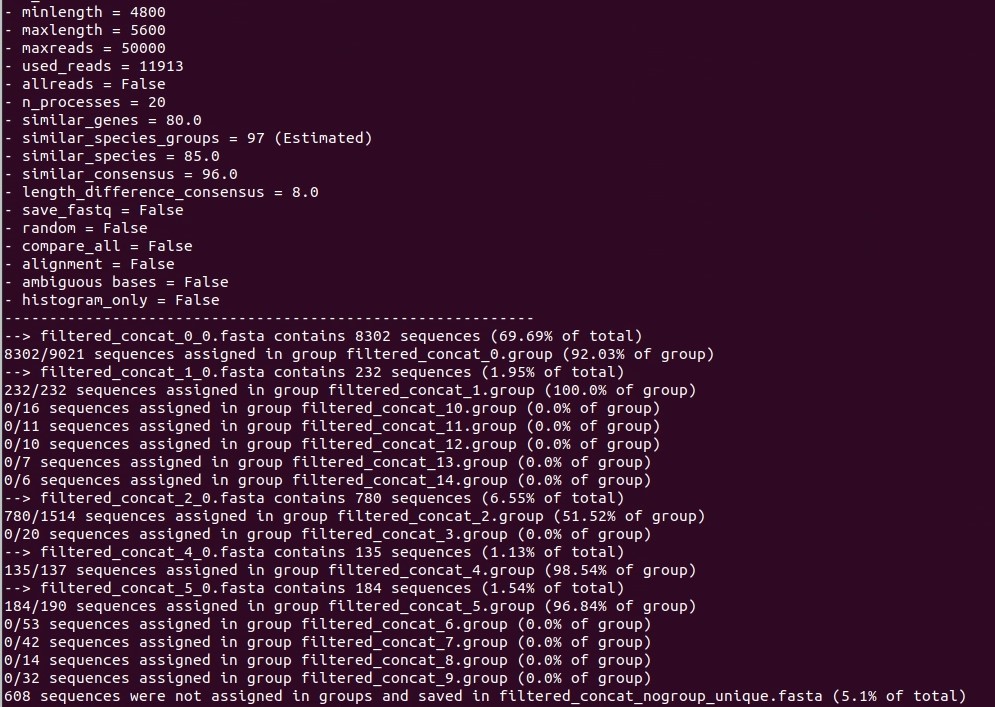
リードがいくつかのグループに分けられ、目的のampliconは、0.groupに振り分けられて8302を使ってconsensus配列が出力される。おそらく正確性に関しては十分すぎるぐらい十分。